Research
Thanks for visiting the Hwang Lab! Our areas of research are
computational biophysics and molecular biomechanics. We perform
computer simulation, modeling, and analysis, to study a range of
biological systems and processes including molecular motors, immune
systems, biofilaments, and macromolecular self-assembly. We also
develop relevant computational and theoretical tools. Our big
question is: How do biomolecules move, interact, and assemble, to
drive life phenomena? Addressing these questions using computers is
both fun and rewarding. Through a better understanding of how life
works, our efforts will also help with developing therapeutic
strategies for various diseases and potentially lead to bio-inspired
engineering.
Projects
Modeling in Multiple Scales
 We study a broad range of problems that are elements of
biological phenomena at different length and time scales. These
studies are closely related to our other projects including
kinesins and T-cell receptors.
We study a broad range of problems that are elements of
biological phenomena at different length and time scales. These
studies are closely related to our other projects including
kinesins and T-cell receptors.
Kinesin
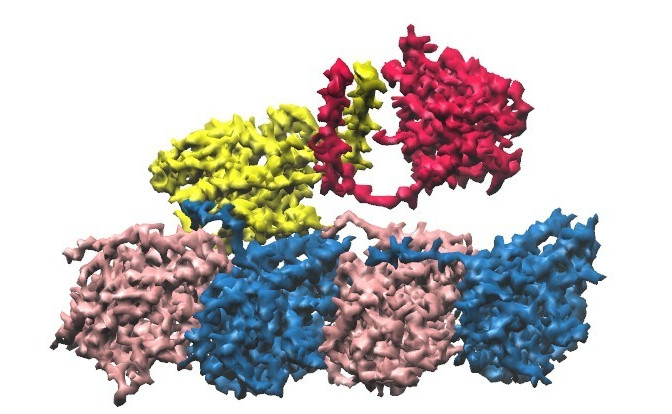 Kinesin is a motor protein that walks along the microtubule filament
in the cell. There are different types of kinesins, specialized for
tasks such as cargo transport or cell division. They exhibit
impressive and diverse motility behaviors. Some kinesins walk like
bipeds; others jump. The microtubule, kinesin's track, has polarity
and its two ends are called plus and minus ends. Kinesins are
unidirectional, walking towards either plus or minus ends, depending
on the kinesin family. There are even kinesin families that
depolymerize microtubules, like running on a bridge simultaneously as
you break it by stomping. How do kinesins do all these?
Kinesin is a motor protein that walks along the microtubule filament
in the cell. There are different types of kinesins, specialized for
tasks such as cargo transport or cell division. They exhibit
impressive and diverse motility behaviors. Some kinesins walk like
bipeds; others jump. The microtubule, kinesin's track, has polarity
and its two ends are called plus and minus ends. Kinesins are
unidirectional, walking towards either plus or minus ends, depending
on the kinesin family. There are even kinesin families that
depolymerize microtubules, like running on a bridge simultaneously as
you break it by stomping. How do kinesins do all these? We use various computational methods, mainly molecular dynamics simulation, to address this question. We study how kinesin processes its fuel molecule (adenosine triphosphate; ATP). Unlike a macroscopic gasoline engine where the energy is is obtained by burning gas, kinesin utilizes energies associated with binding of an ATP molecule (fuel injection), ATP hydrolysis (burning), and the release of hydrolysis products (exhaust) at different phases of its motility cycle. We also investigate how the chemical energy associated with processing ATP is converted to mechanical work, to generate a unidirectional step. Beyond a single kinesin molecule, how a team of kinesins work together to carry cargoes or organize microtubules, so called emergent behaviors, are also of interest.
T-Cell Receptor
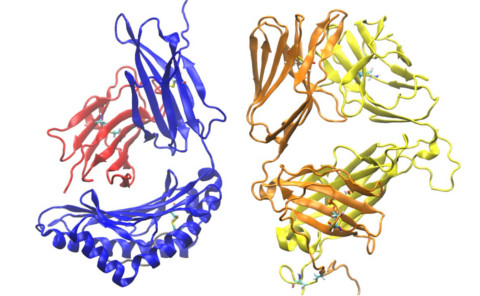 T-cells are major defenders of our body against invaders as well as
against cancerous or damaged cells. Millions of different T-cells
move in our body for immune surveillance. T-cells have exquisite
sensitivity, and fewer than 10 target peptides out of 100,000 present
on the surface of antigen-presenting cell (APC) can be recognized by
a T-cell. Mis-recognition of self-peptide can lead to autoimmune
diseases. Due to its extreme sensitivity, T-cells can also be
utilized to specifically target cancer cells (cancer
immunotherapy). The main molecule on the surface of a T-cell
recognizing antigens is the T-cell receptor (TCR). How a TCR works is
not well understood. An emerging concept is that as a T-cell crawls
over an APC, mechanical load applied to TCR enhances its sensitivity
and the lifetime of the bond to its partner molecule on the surface
of APC, the antigen peptide-bound major histocompatibility complex
(pMHC). We use computer simulation to decipher the physical
mechanisms of this process. Load-dependent conformational changes,
dynamics at the TCR-pMHC interface, and their effect on the peptide
antigen recognition, are being investigated.
T-cells are major defenders of our body against invaders as well as
against cancerous or damaged cells. Millions of different T-cells
move in our body for immune surveillance. T-cells have exquisite
sensitivity, and fewer than 10 target peptides out of 100,000 present
on the surface of antigen-presenting cell (APC) can be recognized by
a T-cell. Mis-recognition of self-peptide can lead to autoimmune
diseases. Due to its extreme sensitivity, T-cells can also be
utilized to specifically target cancer cells (cancer
immunotherapy). The main molecule on the surface of a T-cell
recognizing antigens is the T-cell receptor (TCR). How a TCR works is
not well understood. An emerging concept is that as a T-cell crawls
over an APC, mechanical load applied to TCR enhances its sensitivity
and the lifetime of the bond to its partner molecule on the surface
of APC, the antigen peptide-bound major histocompatibility complex
(pMHC). We use computer simulation to decipher the physical
mechanisms of this process. Load-dependent conformational changes,
dynamics at the TCR-pMHC interface, and their effect on the peptide
antigen recognition, are being investigated.
Bioimage Analysis
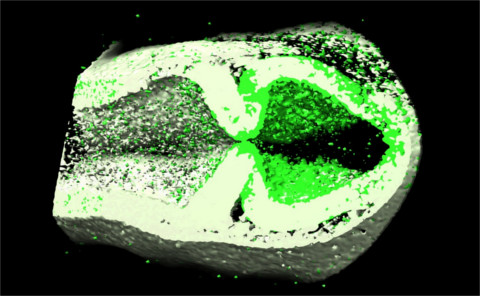 Due to the rapidly advancing imaging technologies, increasingly large
imaging datasets are produced 2-, 3-, and 4-dimensions (3 spatial
dimensions plus time). Yet, information from bioimages are extracted
largely by human eyes, usually assisted by mouse clicking on images
to measure features. Such semi-manual methods become prohibitive as
imaging data are becoming very large. Although a certain level of
automation is possible for systems that exhibit relatively clear
features, more often it is difficult to make the computer to
recognize features in noisy images that human eyes (and brains) can
easily do. Another challenge is image-based model building. For
biomolecular structures, experiments such as x-ray crystallography,
electron microscopy, and nuclear magnetic resonance, provide
atomistic models so that we can perform molecular dynamics
simulations. Can we use the imaging data to build structural models
of, for example, the cytoskeletal network or the mitotic spindle, and
perform meso-scale simulation? To achieve these goals, we are
developing the Computer-Aided Feature Extraction (CAFE)
program. Our present applications of CAFE include 2- and 3-dimensional
filament networks (collagen and the microtubule), and
zebrafish brain morphogenesis.
Due to the rapidly advancing imaging technologies, increasingly large
imaging datasets are produced 2-, 3-, and 4-dimensions (3 spatial
dimensions plus time). Yet, information from bioimages are extracted
largely by human eyes, usually assisted by mouse clicking on images
to measure features. Such semi-manual methods become prohibitive as
imaging data are becoming very large. Although a certain level of
automation is possible for systems that exhibit relatively clear
features, more often it is difficult to make the computer to
recognize features in noisy images that human eyes (and brains) can
easily do. Another challenge is image-based model building. For
biomolecular structures, experiments such as x-ray crystallography,
electron microscopy, and nuclear magnetic resonance, provide
atomistic models so that we can perform molecular dynamics
simulations. Can we use the imaging data to build structural models
of, for example, the cytoskeletal network or the mitotic spindle, and
perform meso-scale simulation? To achieve these goals, we are
developing the Computer-Aided Feature Extraction (CAFE)
program. Our present applications of CAFE include 2- and 3-dimensional
filament networks (collagen and the microtubule), and
zebrafish brain morphogenesis.